Thalassémie : graves conséquences d’une petite mutation
La thalassémie. De quoi s’agit-il?
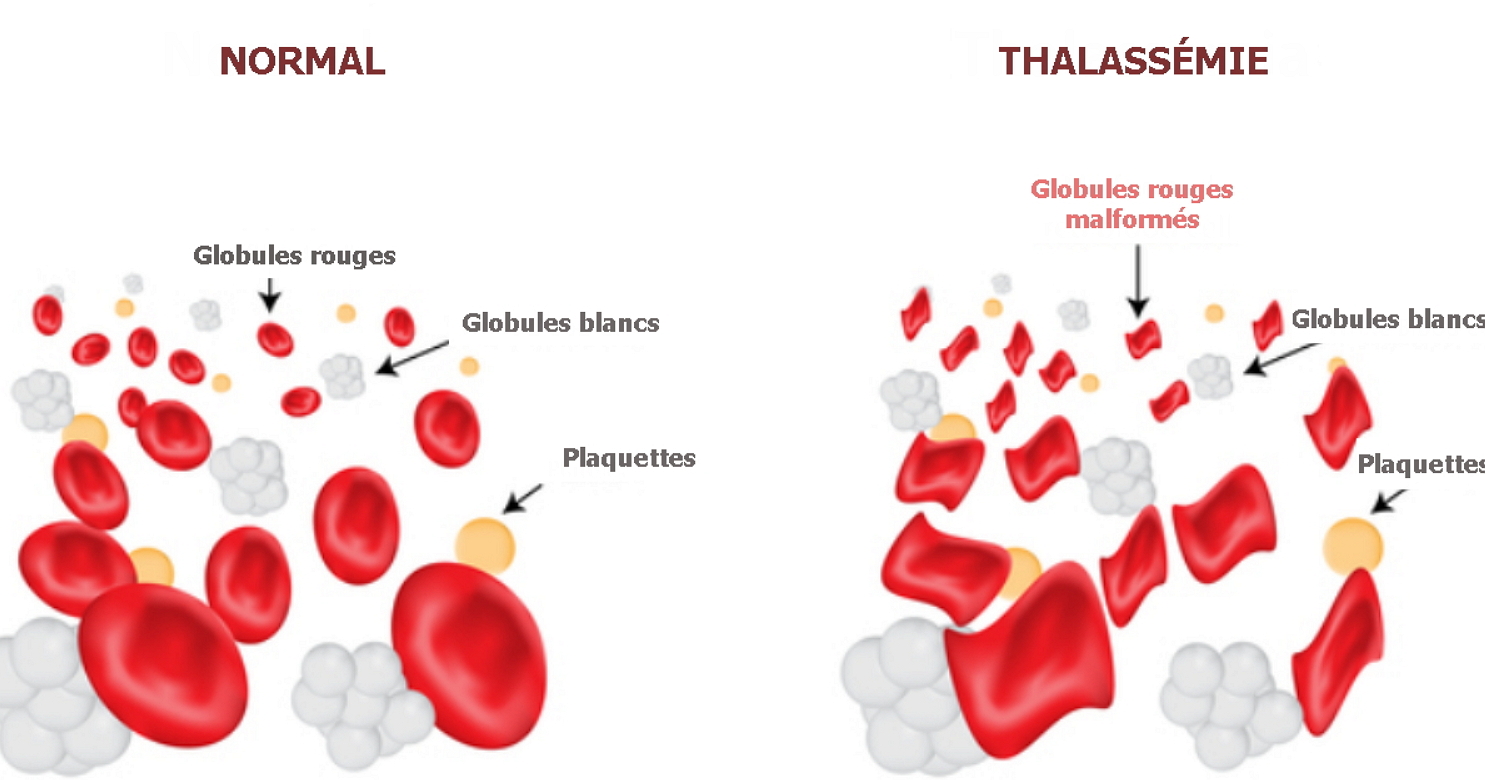
La thalassémie est une maladie héréditaire grave due à une mutation des gènes de l’hémoglobine. Cette protéine remplit les globules rouges et lie l’oxygène pour le transporter dans le sang. Lorsqu’une protéine cesse de remplir sa fonction en raison de mutations, le corps est gravement altéré.
Comment se manifeste la thalassémie ?
- Tout d’abord, la thalassémie se caractérise par une forme sévère d’anémie – une diminution de la quantité d’hémoglobine et d’érythrocytes et, par conséquent, une violation du transfert d’oxygène et de la respiration. En raison de l’anémie, une faiblesse, des étourdissements et un essoufflement se produisent.
- De plus, avec la thalassémie, le corps est souvent surchargé de fer : après tout, il se forme moins d’hémoglobine et les atomes de fer restent sous forme libre dans le sang. Le fer s’accumule dans les organes et les tissus et les empêche de fonctionner correctement. Le foie, le cœur et les glandes endocrines en souffrent.
- Une autre conséquence dangereuse de la thalassémie est l’augmentation de la taille de la rate. C’est dans cet organe que se produit la dégradation des érythrocytes, qui augmente avec la maladie. Plus la rate est volumineuse, plus le risque de rupture est élevé, ce qui met le pronostic vital des patients en danger.
- De plus, avec la thalassémie, les os sont souvent déformés. En effet, en raison du nombre réduit d’érythrocytes dans la moelle osseuse, les zones d’hématopoïèse s’étendent. Les cellules de la moelle osseuse peuvent même s’étendre à l’extérieur des os, formant des épaississements – des pseudotumeurs. Cela conduit non seulement à une déformation du squelette, mais également à d’autres symptômes – après tout, les os peuvent toucher les nerfs et les vaisseaux sanguins.
Mutations de l’hémoglobine et gravité de la maladie
L’hémoglobine se compose de deux types de chaînes – alpha et bêta. Conformément au gène de la chaîne dans laquelle la mutation s’est produite, les thalassémies sont également divisées en types alpha et bêta. Dans ce cas, la chaîne alpha de l’hémoglobine chez l’humaine est codée par deux paires de gènes à la fois, et la chaîne bêta n’en est qu’une.
Selon le nombre d’allèles mutés, les thalassémies sont classées par gravité.
Alpha-thalassémie
Ainsi, si une seule copie du gène de l’alpha-globine cesse de fonctionner correctement, la maladie est presque asymptomatique.
La perturbation du travail de deux copies conduit à une forme bénigne d’alpha-thalassémie.
Trois copies – à des troubles cliniquement prononcés (cette forme est appelée hémoglobinopathie H).
Si les quatre copies du gène de l’alpha-globine ne fonctionnent pas correctement, la mort survient le plus souvent même au stade du fœtus.
Bêta-thalassémie
La bêta-thalassémie se développe de manière similaire.
Si l’une des deux copies du gène code pour une bêta-globine fonctionnelle, la thalassémie est bénigne.
En présence de mutations dans les deux copies du gène qui affectent la production de bêta-globine, une forme grave de la maladie est observée – la thalassémie de Cooley.
Alpha et Béta en même temps
Mais si des mutations se produisent en même temps dans les chaînes alpha et bêta, la formation d’hémoglobine est compensée et la maladie est plus bénigne.
Le fait est que l’effet négatif des mutations dans les gènes des chaînes d’hémoglobine n’est pas seulement une diminution de la quantité de protéines transportant l’oxygène, mais également une diminution de l’équilibre entre les deux types de chaînes. Pour cette raison, une panne du seul gène de l’alpha-globine peut être encore plus dangereuse que celle de deux types de gènes à la fois. Cependant, bien sûr, de telles combinaisons sont extrêmement rares.
Héritage et épidémiologie
Dans le monde, seulement 5% de la population sont porteurs de mutations dans les gènes des chaînes d’hémoglobine, et parmi ceux-ci, un peu moins de 2% des personnes sont sensibles aux symptômes de la maladie. Il convient également de garder à l’esprit que les habitants de différentes régions sont confrontés à la thalassémie avec des fréquences différentes. Tout d’abord, la maladie touche les habitants de la Méditerranée, ainsi que les pays d’Asie centrale et du Sud.
Les porteurs de la mutation peuvent même ne pas être conscients de leur statut – souvent, les dommages à une seule copie du gène ne provoquent pas de symptômes graves. Cependant, les porteurs de la mutation risquent de transmettre la maladie à leurs enfants : si deux porteurs ont un enfant, il peut obtenir les deux copies du gène muté des parents avec une probabilité d’environ 25 %.
Diagnostique de la thalassémie
Le moyen le plus précis de diagnostiquer la thalassémie, quelle que soit sa gravité, consiste à étudier la structure de l’ADN. L’établissement de la séquence d’ADN permettra de détecter les variantes génétiques perturbées et non seulement de prendre des mesures opportunes en cas de maladie, mais également de planifier une grossesse en cas de porteur asymptomatique.
La thalassémie peut également être diagnostiquée à l’aide de tests sanguins : généraux, biochimiques, ainsi que la détermination de la fraction d’hémoglobine dans le sang. Ces études permettent de déterminer le nombre de globules rouges, leur volume, ainsi que la concentration en hémoglobine et les taux de certaines autres substances, afin de déterminer le type de thalassémie en fonction des résultats obtenus.
Si une thalassémie est suspectée, une échographie abdominale est également réalisée pour évaluer la taille de la rate et du foie touchés par la maladie.
Le test génétique Atlas permet d’identifier les variants pathogènes du gène responsable de l’alpha-thalassémie, de détecter les risques liés à la manifestation des symptômes et à la transmission de la maladie aux enfants.
Thérapie et prévention
Pour le traitement des formes sévères de thalassémie, la transfusion sanguine est utilisée – pour augmenter le nombre de globules rouges et restaurer le transport de l’oxygène, ainsi que la chélation – pour éliminer la forme libre du fer, qui n’est pas incorporée dans la « mauvaise » hémoglobine . Pour la bêta-thalassémie sévère, des transfusions sanguines sont fréquemment nécessaires, toutes les 3 à 4 semaines.
Cependant, cette thérapie permet désormais aux patients de mener une vie normale et restaure l’espérance de vie.
Pour les formes légères de thalassémie, les indications sont plus simples – les médecins conseillent de maintenir un mode de vie sain et de bouger davantage. Les activités sportives permettent de maintenir la minéralisation des os du squelette et d’éviter leur déformation.
Conclusion
- La thalassémie est une maladie héréditaire grave due à des mutations des gènes de l’hémoglobine. Elle se caractérise par une faiblesse et des vertiges, des maladies du foie et du cœur, une augmentation de la taille de la rate, ainsi qu’une déformation des os.
- Il existe deux types de maladies – l’alpha et la bêta-thalassémie. En moyenne, dans le monde, les porteurs de mutations caractéristiques des deux espèces sont d’environ 5%.
- La thalassémie est diagnostiquée par des tests sanguins, une échographie et un test génétique.
- Pour le traitement des formes sévères, des transfusions sanguines fréquentes et l’élimination des formes libres du fer sont utilisées. Pour les formes plus légères, le maintien d’un mode de vie sain est suffisant.







